Эпилептические энцефалопатии
Эпилептические энцефалопатии составляют гетерогенную группу заболеваний, для которых характерны частые эпизоды судорог, устойчивость к лечению противоэпилептическими препаратами и существенная задержка психомоторного развития. Патология часто выявляется у детей разных возрастов, в том числе и у новорожденных. Среди наиболее актуальных проблем эпилептической энцефалопатии остается подбор специфической и эффективной терапии, которая смогла бы улучшить качество жизни пациентов.
Этиологические факторы
Причины развития ранней эпилептической энцефалопатии разнообразны. Их можно разделить на генетические и негенетические. К первой группе относятся:
К генетическим причинам эпилептической энцефалопатии относятся мутации в генах, которых насчитывается более 35: STXBP1, KCNQ2, CASK, SCN2A, SLC25A22, STK9, PLCB1 и т. д. Также заболевание может сочетаться с такими хромосомными аномалиями, как синдром Дауна или синдром Ангельмана. По типу наследования эпилептические энцефалопатии могут быть Х-сцепленными доминантными и рецессивными, а также аутосомно-доминантными и аутосомно-рецессивными.
Симптомы эпилептической энцефалопатии
Клиническая картина заболевания зависит от его тяжести, возраста ребенка, наличия или отсутствия дополнительных патологических изменений. Например, ранняя эпилептическая энцефалопатия может проявляться в виде неадекватной реакции ребенка на яркий свет или громкие звуки, повышенный тонус мышц, беспричинный плач и др. По мере взросления ребенка симптомы становятся более отчетливыми. Нарушения мышечного тонуса могут варьироваться от миоклонии и эпилептических спазмов до тяжелых судорог тонического или атонического типа.
В клинической картине обязательно присутствует задержка психомоторного или психоречевого развития, деменция, которая склонна к прогрессированию. Проявления заболевания могут дополняться очаговой неврологической симптоматикой.
Классификация эпилептических энцефалопатий
Классификация заболевания основана на клинике, этиологии, данных электроэнцефалографии и включает в себя 10 отдельных форм:
Данные формы заболевания могут переходить из одной в другую, поэтому специалисты озадачены доработкой классификации. Не исключено, что в ближайшее время она может измениться.
Диагностика эпилептических энцефалопатий
Сегодня существует большое количество методов, с помощью которых можно выявить ранние эпилептические энцефалопатии. Обследование начинается с общего осмотра и проверки рефлексов ребенка. Так как у новорожденных заболевание может сочетаться с метаболическими нарушениями, то дополнительно проводится исследование, направленное на их выявление. Это могут быть как генетические тесты, так и лабораторные анализы.
Характерную картину можно увидеть на электроэнцефалограмме. Для ранней эпилептической энцефалопатии характерно возникновение паттернов «вспышка-подавление», во время которых не прослеживается физиологический ритм. Этот признак обычно регистрируется во время сна, а в состоянии бодрствования может отсутствовать.
Для детей старшего возраста диагностика дополняется психологическими тестами, которые позволяют выявить расстройства психики, мышления, памяти, речи и других функций. Если требуется установить причину эпилептической энцефалопатии и особенности ее наследования, то врач назначит генетические тесты, направленные на поиск мутации в определенных генах. Пройти такое исследование можно в медико-генетическом центре «Геномед».
Энцефалопатия
Энцефалопатия – это собирательное название, включающее однотипные поражения головного мозга, возникающие под действием различных причин. Патологический процесс сопровождается гибелью отдельных нейронов и разрушением связей между ними. При отсутствии медицинской помощи приводит к прогрессирующему ухудшению состояния вплоть до полной деградации личности.
Общие сведения
Энцефалопатия возникает на фоне нарушения метаболизма в клетках головного мозга. Вне зависимости от причины, патология протекает по единому сценарию. Сначала происходит снижение активности нейронов, затем – постепенная их гибель. Очаги дистрофии располагаются по всему головному мозгу, что вызывает разнообразную симптоматику.
Повреждения носят необратимый характер, но при своевременном обращении к специалисту и качественном лечении состояние пациента может улучшиться. Оставшиеся неповрежденными нейроны частично берут на себя функцию погибших клеток, и работа головного мозга значительно улучшается. Если заболевание было застигнуто на ранней стадии, пациент сохраняет полную ясность ума.
Причины
Поражение нервных клеток может возникнуть на фоне воздействия разнообразных патологических факторов.
Врачи выделяют врожденную и приобретенную энцефалопатию. Первая возникает на фоне неправильного течения беременности или родов и, зачастую, развивается еще во время пребывания плода в утробе матери. Ее признаки обнаруживаются сразу после родов или появляются в первые недели жизни. Диагностикой и лечением этого состояния занимаются неонатологи и педиатры.
Приобретенная энцефалопатия встречается уже во взрослом возрасте. Она подразделяется на несколько видов в зависимости от причины гибели нейронов:
В зависимости от скорости развития процесса выделяют энцефалопатию острую и хроническую. Первая может развиться в течение нескольких дней или часов, чаще возникает на фоне сильной интоксикации, травмы, инфекционного процесса. Хронический процесс может протекать годами и десятилетиями.
Степени
Границы между степенями тяжести энцефалопатии условны, но для удобства врачи пользуются следующей классификацией:
Симптомы
Признаки энцефалоптии зависят от локализации очага разрушения, а также степени развития заболевания. Наиболее часто пациенты и их родственники сталкиваются со следующими симптомами:
У одних пациентов преобладают расстройства поведения, другие перестают нормально владеть своим телом; у третьих страдают, в основном, органы чувств. В тяжелых случаях человек требует постоянного ухода и наблюдения окружающих.
Осложнения
Осложнения энцефалопатии связаны с сильным и необратимым поражением головного мозга и представляют собой предельную степень наблюдаемых симптомов:
Диагностика
Диагностикой и лечением энцефалопатии занимается врач невролог. Обследование пациента включает в себя:
При необходимости могут быть назначены другие анализы и обследования, а также консультации специалистов для точного определения причины энцефалопатии.
Лечение
Чем раньше будет начато лечение энцефалопатии, тем больше шансов остановить прогрессирование патологии и восстановить полноценную работу головного мозга.
В первую очередь, необходимо устранить причину поражения нейронов:
Лечение непосредственно энцефалопатии требует назначения препаратов, улучшающих кровоток в сосудах головного мозга и обмен веществ внутри клетки. В зависимости от причины и степени поражения, врачи могут назначить:
При энцефалопатии курсы лечения проводятся регулярно, минимум 2 раза в год. Это позволяет держать заболевание под контролем. Подбор конкретных препаратов и определение дозировки осуществляется только врачом. Единой схемы лечения для всех больных не существует.
Для усиления действия лекарственных препаратов используются немедикаментозные методы лечения:
В отдельных случаях (при сосудистой природе энцефалопатии) приходится прибегать к хирургическому лечению:
Профилактика
Энцефалопатия – сложное заболевание. Не существует четкой методики, позволяющей предупредить ее появление и устранить все факторы риска. Врачи рекомендуют придерживаться следующих правил:
Лечение в клинике «Энергия здоровья»
Врачи клиники «Энергия здоровья» всегда готовы принять пациента любого возраста. Мы проведем тщательное обследование, выявим возможные причины энцефалопатии и примем все меры по ее устранению:
Наши неврологи будут контролировать состояние пациента и корректировать лечение при необходимости. Лечение энцефалопатии – это длительный и сложный процесс, но мы готовы за него взяться.
Преимущества клиники
Если Вам требуется полноценное обследование и качественное лечение, добро пожаловать в клинику «Энергия здоровья». Здесь Вас ждут:
Если у Вас или Ваших родственников появились признаки энцефалопатии, не затягивайте с обращением к врачу. Чем будет начато лечение, тем лучше результат. Запишитесь в клинику «Энергия здоровья» и позвольте нашим врачам подобрать оптимальную терапию.
Эпилепсия в детском возрасте
Эпилепсия — общее название группы хронических пароксизмальных болезней головного мозга, проявляющихся повторными судорожными или другими (бессудорожными) стереотипными припадками, сопровождающихся разнообразными (патологическими) изменениями личности и сн
Часть 3. Начало статьи читайте в № 6, 8, 2014 год
Существует немало форм эпилепсии, встречающихся исключительно в детском или подростковом возрасте. Именно зависимость от возраста многих разновидностей эпилепсии является главным отличительным признаком эпилептологии детского возраста [1–4].
Эпилепсии и судорожные синдромы периода новорожденности
Хотя продолжительность неонатального периода невелика, целый ряд эпилептических синдромов свойственен именно для новорожденных детей [3–5].
Доброкачественные семейные приступы (судороги) новорожденных
Доброкачественная неонатальная эпилепсия (с аутосомно-доминатным типом наследования) трех типов, проявляющаяся в первые 7 дней жизни (начиная с трех суток). В семейном анамнезе обязательно фигурируют указания на наличие в прошлом судорог у членов семьи пациента (в неонатальном периоде). Связь припадков с уточненными врожденными нарушениями метаболизма не установлена. Доброкачественные семейные неонатальные приступы манифестируют в виде фокальных и мультифокальных или генерализованных тонико-клонических (судорожных) припадков. Указанные припадки характеризуются малой продолжительность (1–2 мин) и значительной частотой (20–30 эпизодов за сутки). Впоследствии, по прошествии от 1 до 3 недель, приступы самопроизвольно спонтанно купируются.
Доброкачественные несемейные судороги (приступы) новорожденных («припадки пятого дня»)
Эта эпилепсия с дебютом в раннем неонатальном периоде имеет также другое название (доброкачественные идиопатические неонатальные судороги). Болезнь впервые описана в конце 1970-х гг. Судорожные приступы развиваются у доношенных новорожденных детей, не имевших до этого признаков патологии со стороны ЦНС. Дебют приступов происходит к концу 1-й недели жизни (в 80–90% случаев — между 4-м и 6-м днями), а их пик приходится на 5-й день жизни (отсюда и название). Описываемые приступы обычно имеют вид мультифокальных клонических судорог, которым нередко сопутствуют апноэ. В большинстве случаев доброкачественные идиопатические неонатальные судороги длятся не более 24 ч (они всегда прекращаются по прошествии 15 дней после дебюта). В 80% случаев за время судорожного периода у новорожденных отмечается развитие эпилептического статуса [3–5].
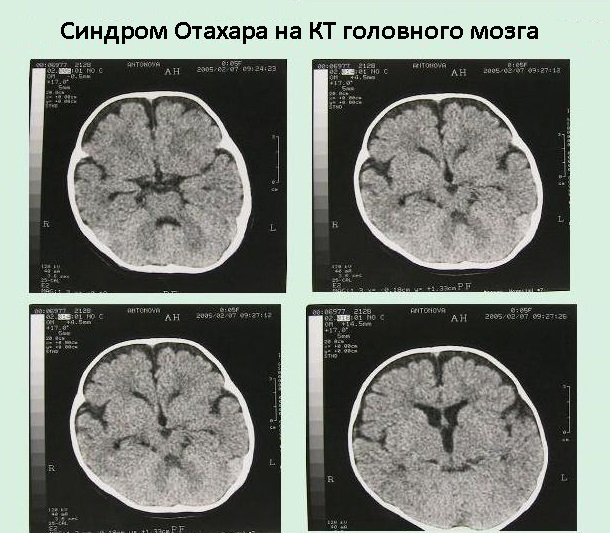
Ранняя инфантильная эпилептическая энцефалопатия с паттерном «угнетение/вспышка» на ЭЭГ (синдром Отахары)
Ранняя инфантильная эпилептическая энцефалопатия — редкая болезнь, относящаяся к злокачественным формам эпилепсии детского возраста. Дебютирует обычно в периоде новорожденности (или в возрасте 1–3 мес). Болезнь характеризуется тоническими приступами, частота которых значительно варьирует (10–300 эпизодов за сутки). У детей отмечается быстрое формирование неврологического дефицита и задержка психического развития. Специфический паттерн «вспышка/угнетение» при электроэнцефалографии (ЭЭГ) представлен у детей c синдромом Отахары как в состоянии сна, так и при бодрствовании. При магнитно-резонансной томографии (МРТ) головного мозга у пациентов отмечаются грубые аномалии развития ЦНС. Среди детей с ранней инфантильной эпилептической энцефалопатией с паттерном «вспышка/угнетение» на ЭЭГ летальность к возрасту 1 года достигает 40–50%. В 4–6-месячном возрасте синдром Отахары может трансформироваться в синдром Веста [3–6].
Ранняя миоклоническая (эпилептическая) энцефалопатия
Описана J. Aicardi и F. Goutières (1978); дебютирует преимущественно в периоде новорожденности (иногда до 3-месячного возраста). В генезе болезни предполагается роль генетических факторов и некоторых «врожденных ошибок метаболизма» (пропионовая ацидурия, метилмалоновая ацидемия, болезнь мочи с запахом кленового сиропа и др.). Клинически проявляется частыми миоклоническими припадками. Последние обычно не ассоциированы с ЭЭГ-изменениями во время приступа, но в ряде случаев одновременно с миоклониями регистрируются эпилептиформные разряды «угнетение/вспышка». Миоклонии чаще бывают фрагментарными (легкие подергивания дистальных отделов конечностей, век или углов рта); одновременно могут отмечаться фокальные (парциальные) приступы, массивные миоклонии и тонические спазмы (изолированные или серийные — возникают к 3–4 месяцам). Появление у ребенка тонических спазмов заставляет предположить наличие атипичного синдрома Веста, но вскоре основные проявления болезни возобновляются и сохраняются на протяжении длительного времени. Фокальные припадки (сложные парциальные — с заведением глаз или автономными симптомами: апноэ, гиперемия лица; клонические судороги в разных участках тела и др.) становятся основным типом приступов при ранней миоклонической эпилептической энцефалопатии. При интериктальном ЭЭГ-исследовании у детей регистрируется паттерн «угнетение/вспышка», состоящий из разрядов продолжительностью 1–5 сек, чередующийся с почти изоэлектрическими периодами (длительностью 3–10 сек). Описываемый ЭЭГ-паттерн становится более отчетливым во время сна (особенно в фазе глубокого сна). Изначальный паттерн «угнетение/вспышка» по достижении возраста 3–5 мес сменяется атипичной гипсаритмией или мультифокальными пароксизмами, но в большинстве случаев это лишь транзиторный феномен. Болезнь сопровождается высокой летальностью или прогрессивным распадом психомоторных функций (вплоть до вегетативного статуса), хотя по мере увеличения возраста частота и выраженность фокальных приступов и миоклоний постепенно уменьшаются [3–5, 7].
Витамин В6-зависимая эпилепсия
Cравнительно редкое наследственное заболевание, характеризующееся фармакорезистентными судорогами. Относится к группе метаболически обусловленных эпилепсий. Развивается у новорожденных, матери которых длительно получали пиридоксин во время беременности, а также при специфическом наследственном дефекте метаболизма (с повышенной потребностью в витамине В6). Известны случаи дебюта пиридоксинзависимых судорог у детей старше 1 мес и даже на втором году жизни. Между приступами судорог дети остаются беспокойными, реагируют мышечными подергиваниями на внешние раздражения. Болезнь не поддается обычному противосудорожному лечению, но назначение витамина В6 в высоких дозах (25 мг/кг/сут) быстро приводит к нормализации состояния [3–5].
Злокачественные мигрирующие парциальные судороги (приступы) младенческого возраста
Чрезвычайно редко встречающийся эпилептический синдром, описанный G. Coppola и соавт. (1995). К настоящему времени сообщается всего о примерно 50 случаях болезни, зарегистрированных в различных странах мира. Злокачественные мигрирующие парциальные судороги в 50% случаев наблюдаются в первые дни жизни; остальные 50% приходятся на возраст 1–3 мес. При дебюте приступы носят фокальный клонический характер, а по прошествии нескольких недель они становятся мультифокальными, причем исключительно частыми и фармакорезистентными к терапии антиэпилептическими препаратами. При ЭЭГ-исследовании у детей выявляется выраженная многоочаговая эпилептическая активность; метаболических нарушений не обнаруживается, а МРТ-признаки патологических изменений отсутствуют. Паталогоанатомическое исследование позволило выявить в гиппокампе признаки нейрональной потери [1, 3, 5, 8].
Эпилепсии у детей первого года жизни (1–12 мес)
По достижении 1-месячного возраста число разновидностей эпилептических синдромов, специфичных для первого года жизни ребенка, практически не уступает таковому, свойственному периоду новорожденности.
Инфантильные спазмы (синдром Веста)
Этот вариант катастрофической эпилепсии (генерализованной) бывает симптоматическим (подавляющее большинство случаев) или криптогенным (10–20%). Он манифестирует у детей на первом году жизни (чаще между 3-м и 8-м месяцами). В классическом варианте синдром Веста характеризуется в момент приступа комбинацией сгибательных и разгибательных движений, то есть выраженными миоклоническими (салаамовыми) спазмами, иногда серийными короткими сгибательными движениями головы («кивки»). Инфантильные спазмы могут развиться как вследствие наличия различной неврологической патологии, так и без каких-либо очевидных предшествующих нарушений функций ЦНС. При инфантильных спазмах психомоторное развитие замедляется, в дальнейшем высока вероятность выраженного отставания в развитии. В 80% случаев при синдроме Веста обнаруживаются микроцефалия, признаки детского церебрального паралича, атонически-атактические нарушения и др. Отличительным электрофизиологическим признаком синдрома Веста является гипсаритмия (по данным ЭЭГ), которая имеет вид диффузных высоковольтажных пиков и медленных волн, располагающихся на дезорганизованном (медленном) фоне. Прогноз синдрома Веста определяется эффективностью проводимой терапии, но в целом малоблагоприятен [3–8].
Тяжелая миоклонус-эпилепсия младенческого возраста (синдром Драве)
Болезнь, описанная C. Dravet (1978, 1992), дебютирует на первом году жизни (между 2-м и 9-м мес), что нередко происходит вслед за развитием фебрильного эпизода, вскоре после вакцинации или перенесения инфекции. Синдром Драве характеризуется появлением генерализованных или односторонних клонических судорог (обычно на фоне гипертермии или лихорадки), что происходит на фоне предшествующего нормального психомоторного развития ребенка на протяжении первого года жизни. Постепенно (по прошествии нескольких недель или месяцев) у ребенка развиваются афебрильные миоклонические и парциальные (фокальные) припадки. Прогрессивное нарастание частоты миоклоний (изолированных или серийных) предшествует появлению у пациентов генерализованных припадков. У детей выявляются умеренные мозжечковые и пирамидные знаки, связанные с дефицитарностью грубой моторики и атаксией походки. Нарушения психомоторного развития впоследствии отмечаются у детей примерно до 4-летнего возраста. Нередко при синдроме Драве у детей развивается эпилептический статус (судорожный или миоклонический). Данные ЭЭГ на протяжении первого года жизни обычно остаются в пределах нормы, хотя у отдельных пациентов встречаются спонтанные фотоиндуцированные пик-волновые разряды. Впоследствии иктальные ЭЭГ-исследования при синдроме Драве характеризуются наличием миоклонических или клонических припадков (генерализованная пик-волновая или полипик-волновая активность). Генерализованные разряды усиливаются в состоянии релаксации; одновременно отмечаются фокальные и мультифокальные пики и острые волны. Традиционные и новые антиэпилептические препараты обычно не предотвращают рецидива приступов при синдроме Драве. Прогноз по интеллектуальному развитию при синдроме Драве всегда неблагоприятен [3–5, 8].
Идиопатические доброкачественные парциальные эпилепсии младенчества
Обычно дебютируют у детей в возрасте 3–20 месяцев (чаще между 5-м и 8-м мес). Впервые описаны K. Watanabe и соавт. (1987), вследствие чего изначально получили обобщающее название «синдром Ватанабе». Характеризуются проявлениями в виде сложных парциальных (фокальных) приступов и благоприятным прогнозом (элиминация эпилептических припадков в течение 3 мес после дебюта). В среднем число приступов составляет около 7; у части пациентов отмечаются исключительно сложные парциальные припадки, у других — только вторично-генерализованные, а примерно в половине случаев встречается их сочетание. Во время приступа для пациентов характерны снижение реакции на предъявляемые стимулы, остановка двигательной активности, умеренные судорожные подергивания, латеральное заведение глаз и цианоз. Основными клиническими признаками этой группы эпилепсий являются высокая встречаемость кластерных приступов, короткая продолжительность припадков, а также изначально нормальные показатели интериктального ЭЭГ-исследования (впоследствии у части детей могут обнаруживаться пароксизмальные разряды) [2, 3, 5, 6, 8].
Сходные с идиопатическими доброкачественными парциальными эпилепсиями младенчества, но исключительно семейные пароксизмальные состояния с дебютом на первом году жизни носят название «доброкачественные инфантильные семейные судороги». В 1997 г. были описаны сходные с ними случаи семейных эпилепсий с последующим формированием хореоатетоза — семейные судороги с хореатетозом [3–5, 8, 9].
Эпилепсии у детей раннего возраста (1–3 года)
Для детей раннего возраста (от 12 до 36 месяцев), в первую очередь, характерны cиндром Доозе, синдром Леннокса–Гасто, доброкачественная миоклонуc-эпилепсия младенческого возраста, синдром гемиконвульсий-гемиплегии, идиопатическая парциальная эпилепсия младенчества, абсансная эпилепсия раннего детства, электрический эпилептический статус медленно-волнового сна, ранний и поздний детский нейрональный липофусциноз (типы I и II).
Миоклоническая астатическая эпилепсия раннего детского возраста (cиндром Доозе)
Представляет собой эпилепсию c миоклонически-астатическими приступами (различной продолжительности). Приступы дебютируют в возрасте 1–5 лет. Чаще болезнь поражает мальчиков. Астатические и миоклонические приступы могут сочетаться, причем миоклонии возникают как до, во время, так и после астатического припадка. Приступы наступают внезапно и практически всегда сопровождаются падениями. Миоклонии отмечаются в виде различной выраженности симметричных подергиваний в руках и мышцах плеч пояса, что сочетается с наклоном головы («кивки»). Признаки утраты сознания у детей в момент приступа отсутствуют. До начала заболевания психомоторное развитие детей обычно соответствует норме. У части детей болезнь осложняется риском развития деменции (предположительно за счет развития эпилептического статуса абсансов). При ЭЭГ регистрируются генерализованные билатерально-синхронные комплексы пик-волн (3 и более за 1 сек, 2–4 Гц). Прогноз при миоклонически-астатической эпилепсии раннего детского возраста малоблагоприятен [3–6, 8].
Синдром Леннокса–Гасто, или миокинетическая эпилепсия раннего детства с медленными пик-волнами
Группа гетерогенной патологии с эпилептическими приступами (атоническими, тоническими, атипичными абсансами), интеллектуальной дефицитарностью и характерным ЭЭГ-паттерном. Как и при синдроме Веста, при синдроме Леннокса–Гасто выделяют симптоматический и криптогенный варианты болезни. Ранние формы дебютируют примерно с 2-летнего возраста. До 30% случаев представляют собой результат трансформации из синдрома Веста. Клинически синдром Леннокса–Гасто характеризуется миоклонически-астатическими припадками, салаамовыми судорогами (молниеносными кивательными), атипичными абсансами, тоническими приступами (чаще во сне). Могут встречаться генерализованные тонико-клонические, миоклонические и фокальные (парциальные) приступы. Для детей типична серийность припадков с изменениями сознания (ступор) и постепенным переходом в эпилептический статус. Помимо эпилептических приступов, в неврологическом статусе могут отмечаться церебральные парезы/параличи, а также атонически-астатические нарушения (до 40% пациентов). У детей происходит снижение интеллекта (различной степени), наблюдаются выраженные нарушения когнитивных функций. По ЭЭГ-данным типичны изменения фоновой активности в виде медленных пик-волн
В. М. Студеникин, доктор медицинских наук, профессор, академик РАЕ
ФГБУ «НЦЗД» РАМН, Москва
Что такое эпилептическая энцелофапатия
Отдел психоневрологии и эпилептологии Научно-исследовательского клинического института педиатрии им. Ю.Е. Вельтищева ФГБУ ВО «Российский национальный исследовательский медицинский университет им. Н.Н. Пирогова» Минздрава России, Москва, Россия
Московский НИИ педиатрии и детской хирургии
ФГБУ Медико-генетический научный центр РАМН, Москва
Генетика и дифференцированное лечение ранних эпилептических энцефалопатий
Журнал: Журнал неврологии и психиатрии им. С.С. Корсакова. Спецвыпуски. 2016;116(9-2): 67-73
Шарков А. А., Шаркова И. В., Белоусова Е. Д., Дадали Е. Л. Генетика и дифференцированное лечение ранних эпилептических энцефалопатий. Журнал неврологии и психиатрии им. С.С. Корсакова. Спецвыпуски. 2016;116(9-2):67-73.
Sharkov A A, Sharkova I V, Belousova E D, Dadali E L. Genetics and treatment of early infantile epileptic encephalopathies. Zhurnal Nevrologii i Psikhiatrii imeni S.S. Korsakova. 2016;116(9-2):67-73.
https://doi.org/10.17116/jnevro20161169267-73
Отдел психоневрологии и эпилептологии Научно-исследовательского клинического института педиатрии им. Ю.Е. Вельтищева ФГБУ ВО «Российский национальный исследовательский медицинский университет им. Н.Н. Пирогова» Минздрава России, Москва, Россия

Эпилептические энцефалопатии (ЭЭ) — группа прогрессирующих заболеваний различной этиологии, которые проявляются нейрокогнитивным дефицитом и эпилептиформной активностью на электроэнцефалограмме. ЭЭ составляют около 15% от всех форм эпилепсии детского возраста и до 40% от всех эпилептических приступов, возникающих в первые 3 года жизни. Выделяют 10 синдромальных форм ЭЭ. Генетические факторы играют определенную роль в развитии патологии примерно у 70—80% пациентов, и не менее 40% всех идиопатических эпилепсий имеют моногенную природу. Идентифицировано 35 генов, ответственных за возникновение ЭЭ, и их поиск продолжается. Показана выраженная генетическая гетерогенность ранних ЭЭ, 16 из которых наследуются аутосомно-доминантно, 13 — аутосомно-рецессивно, 4 — Х-сцепленно рецессивно и 2 — Х-сцепленно доминантно. Приведены дифференцированные подходы к лечению отдельных синдромов ЭЭ. В публикациях последних лет показана эффективность таргетной терапии при некоторых моногенных формах ранних ЭЭ (стирипентола при мутациях в гене SCN1A, дифенина при мутациях в гене SCN8A, леветирацетама при мутациях в гене STXBP1). Полученные результаты исследований свидетельствуют о необходимости проведения точной диагностики генетического варианта ранних ЭЭ, что позволит не только осуществить профилактические мероприятия в отягощенной семье, но и повысить эффективность лечения.
Отдел психоневрологии и эпилептологии Научно-исследовательского клинического института педиатрии им. Ю.Е. Вельтищева ФГБУ ВО «Российский национальный исследовательский медицинский университет им. Н.Н. Пирогова» Минздрава России, Москва, Россия
Московский НИИ педиатрии и детской хирургии
ФГБУ Медико-генетический научный центр РАМН, Москва
Термин «эпилептическая энцефалопатия» (ЭЭ) используется для обозначения гетерогенной группы заболеваний, характеризующихся частыми полиморфными судорожными пароксизмами, резистентными к противосудорожной терапии, и «агрессивной» межприступной эпилептиформной активностью, ассоциированной с выраженной задержкой психомоторного развития.
Первое описание заболевания из этой группы было сделано английским врачом W. West, который в 1841 г. представил полный анализ особенности клинических проявлений на примере собственного ребенка. Выделение этих состояний в самостоятельную группу заболеваний продолжил H. Gastaut в 1964 г. Он предложил в качестве обязательного симптома ЭЭ прогрессирующую умственную отсталость, считая ее следствием межприступной эпилептической активности на электроэнцефалограмме (ЭЭГ). На связь нейропсихологического дефицита и непрерывной или преходящей межприступной пароксизмальной активности на ЭЭГ указывали W. Landau и F. Kleffner в первом описании одноименного синдрома еще в 1957 г.
Современная концепция ЭЭ представлена в работах O. Dulac в 90-х годах (цит. по [1]). Она нашла отражение в классификации Международной Лиги по борьбе с эпилепсией (ILAE) в 2001 г., в которой термин «эпилептическая энцефалопатия» был использован для определения «эпилептиформных изменений, способствующих прогрессивному нарушению функций мозга» [1].
В 2006 г. J. Engel [2] определяет ЭЭ как состояния, «в основе нейродегенеративных изменений или психомоторной задержки которых лежит сама эпилепсия», в отличие от лежащих в основе других синдромов метаболических, дегенеративных или энцефалитических процессов, тем самым исключая эти конкретные прогрессирующие патологии из этиологического спектра Э.Э. Однако впоследствии аргументы в пользу пересмотра этой концепции и включения в эту группу различных по этиологии заболеваний были сформулированы G. Capovilla и соавт. (цит. по [3]). Они предложили новое определение эпилептических энцефалопатий — «прогрессирующие состояния различной этиологии, которые могут вызывать неврологический дефицит как сами по себе, так и за счет эпилепсии». В соответствии с этим определением в группу ЭЭ включается широкий круг неврологической патологии: опухоли головного мозга, нейродегенеративные (например, полидистрофии, лейкодистрофии) или метаболические (например, фенилкетонурия, недостаточность биотинидазы) заболевания, а также воспалительные или аутоиммунные состояния (например, разрушительная эпилептическая энцефалопатия у детей школьного возраста, вызванная гипертермией, и синдром Расмуссена). В публикациях последних лет также отмечается увеличение доли моногенных вариантов в структуре ЭЭ, обусловленных мутациями в генах, продукты которых участвуют в формировании ионных или лигандзависимых каналов нейронов коры головного мозга или осуществляющих биохимические процессы в нейрональных структурах.
Распространенность ЭЭ
Распространенность ЭЭ окончательно не установлена. В немногочисленных работах зарубежных авторов имеются указания только на долю ЭЭ в структуре заболеваний, сопровождающихся судорожным синдромом в детском возрасте. По данным U. Kramer и соавт. [4] и S. Gürsoy и D. Ercal [5], ЭЭ составляют около 15% от всех форм эпилепсии детского возраста и около 40% от всех судорог, возникающих в первые 3 года жизни. В отечественных публикациях [6] эта цифра меньше — около 7% от всех форм эпилепсии до 18 лет. Наиболее часто диагностируются ранние ЭЭ, возникающие в неонатальном или раннем детском возрасте [7].
Особенности клинических проявлений и классификация ЭЭ
Наиболее распространенная клиническая классификация ЭЭ, представленная C. Panayiotopoulos в 2005 г. [9] и модифицированная рядом авторов [10, 11], включает в себя 10 следующих синдромальных форм:
1. Ранняя миоклоническая энцефалопатия.
5. Синдром Леннокса—Гасто.
6. Синдром Ландау—Клеффнера.
7. Синдромы с продолженной спайк-волновой активностью во сне (кроме синдрома Ландау—Клеффнера).
8. Миоклонический статус непрогрессирующих энцефалопатий.
9. Злокачественная эпилепсия детства с мигрирующими парциальными судорогами.
В основу предложенной систематики ЭЭ положены особенности клинических проявлений и ЭЭГ-картины. Но важно, что в каждую из выделяемых групп входят заболевания, имеющие различную этиологию. Наиболее частые причины приведены в табл. 1 [12, 13].
 Таблица 1. Этиология ЭЭ
Таблица 1. Этиология ЭЭ
Как видно из таблицы, одним из этиологических факторов у части больных являются мутации в одном или нескольких генах, что предполагает их значительную роль в возникновении ЭЭ.
В ряде публикаций намечены границы между отдельными Э.Э. Так, в одной из работ Y. Yamatogi и S. Ohtahara [16] было показано, что больше половины случаев синдрома Отахара трансформируется в синдром Веста. В свою очередь синдром Веста может эволюционировать в мультифокальную эпилепсию или синдром Леннокса—Гасто, что дает повод задуматься о необходимости пересмотра классификации ЭЭ, в основу которой будут положены не только особенности клинических проявлений, но и различия в этиологии этих синдромов.
Наследственные варианты ЭЭ
В последние годы значительно возрос интерес к изучению генетических причин Э.Э. Предполагается, что генетические факторы играют определенную роль примерно у 70—80% пациентов с эпилепсией [17], и не менее 40% всех идиопатических эпилепсий имеют моногенную природу [18].
Клинические проявления ЭЭ наблюдаются у больных с ранними ЭЭ, наследственными болезнями обмена (в том числе лизосомными, пероксисомными, митохондриальными, нарушением гликозилирования и др.), а также нейродегенеративными заболеваниями и различными пороками развития мозга (кортикальными дисплазиями, лисэнцефалиями, голопрозэнцефалиями). Всего к настоящему времени в каталог OMIM включено более 400 генов, мутации в которых приводят к возникновению моногенных заболеваний, сопровождающихся судорогами. Кроме того, судорожный синдром входит в симптомокомплекс значительного числа хромосомных синдромов, диагностируемых с помощью как стандартного кариотипа, так и хромосомного микроматричного анализа.
Изолированные варианты наследственных ЭЭ, при которых судорожный синдром долгое время является единственным симптомом, составляют так называемые ранние ЭЭ. К настоящему времени идентифицировано 35 генов, ответственных за их возникновение, и их поиск продолжается. Показана выраженная генетическая гетерогенность ранних ЭЭ, 16 из которых наследуются аутосомно-доминантно, 13 — аутосомно-рецессивно, 4 — Х-сцепленно рецессивно и 2 — Х-сцепленно доминантно (табл. 2).
 Таблица 2. Моногенные ЭЭ Примечание. АР — аутосомно-рецессивный; АД — аутосомно-доминантный; ГТФаза — гуанозинтрифосфат гидролаза, ГФИ — гликозилфосфатидилинозитол, NMDA — N-метил-D-аспартатный рецептор.
Таблица 2. Моногенные ЭЭ Примечание. АР — аутосомно-рецессивный; АД — аутосомно-доминантный; ГТФаза — гуанозинтрифосфат гидролаза, ГФИ — гликозилфосфатидилинозитол, NMDA — N-метил-D-аспартатный рецептор.
Таблица 2. (окончание) Моногенные ЭЭ Примечание. АР — аутосомно-рецессивный; АД — аутосомно-доминантный; ГТФаза — гуанозинтрифосфат гидролаза, ГФИ — гликозилфосфатидилинозитол, NMDA — N-метил-D-аспартатный рецептор.
Наиболее часто диагностируются ранняя ЭЭ 6-го типа (синдром Драве), обусловленная мутациями в гене SCN1A, в то время как другие некоторые генетические варианты описаны лишь в единичных семьях. Кроме того, описано существование нескольких аллельных вариантов ЭЭ, возникающих при мутациях в одном и том же гене. Так, например, мутации в гене SCN1A могут приводить к возникновению не только cиндрома Драве, но и фебрильных судорог 3А-типа, гемиплегической мигрени и эпилепсии из группы генерализованных эпилепсий с фебрильными судорогами плюс [19]. C другой стороны, у больных с клиническими проявлениями синдрома Драве обнаруживаются мутации и в других генах: SСN9A, PCDH19, GABRG2. Обсуждение клинических проявлений каждого варианта ранних ЭЭ выходит за рамки данной статьи.
Наличие выраженной генетической гетерогенности сходных по клиническим проявлениям заболеваний существенно затрудняет диагностику определенного генетического варианта, особенно при наличии единственного больного в семье. Для оптимизации диагностики создаются диагностические алгоритмы, в основу которых положены такие критерии, как тип наследования, частота генетического варианта, наличие мажорных мутаций в гене, ответственном за его возникновение, существование специфических биохимических маркеров и особенности клинических проявлений. При существовании предположения о том, что заболевание обусловлено нарушением обмена веществ, в диагностический алгоритм включают исследование биохимических маркеров при проведении тандемной масс-спектрометрии, высокожидкостной газовой хроматографии органических кислот и определении концентрации трансферринов и ферритинов в крови. Однако использование этих методов целесообразно лишь в тех случаях, когда с высокой долей вероятности предполагается наличие у больного одного из вариантов наследственной болезни обмена веществ, а для идентификации генетического варианта с использованием молекулярно-генетических методов не обязательно. В тех случаях, когда идентификация гена, мутации в котором привели к заболеванию, затруднена, целесообразнее использовать самые современные методы молекулярного анализа, такие как высокопроизводительное секвенирование экзома нового поколения. Использование этого метода позволяет одновременно тестировать мутации в нескольких сотнях или даже тысячах генов, что позволяет существенно увеличить эффективность диагностики и снизить экономические затраты на ее проведение.
Лечение
Из табл. 2 видно, что многие распространенные моногенные генетические варианты ЭЭ обусловлены мутациями в генах, кодирующих потенциал- и лигандзависимые каналы нейронов, а также ферменты и никотиновые холиновые рецепторы, функционирование которых обеспечивает прохождение нервного импульса в нейронах коры головного мозга. Известно, что терапевтический эффект большинства противоэпилептических препаратов (ПЭП) основан на модуляции каналов нейронов коры головного мозга, усилении тормозной синаптической передачи или торможении активирующей синаптической передачи. Однако точный механизм того, как это препятствует развитию судорог, известен не для всех ПЭП. Выявлено, что фенитоин, ламотриджин и препараты из группы карбамазепина влияют на быструю иннактивацию натриевых каналов, в то время как лакосамид — на медленную. Топирамат и фелбамат влияют на функционирование ГАМК-зависимых каналов [20]. Блокаторами низковольтажных кальциевых каналов являются этосуксимид, а высоковольтажных — габапентин и прегабалин. Особенно интересно ГАМКергическое влияние некоторых препаратов: пролонгирование открытия хлорных каналов у барбитуратов и учащения открывания у бензодиазепинов; ингибирование ГАМК-трансаминазы вигабатрином, блокирование обратного захвата ГАМК в синапсе тиагабином [21].
Однако при выборе терапевтической стратегии необходимо иметь в виду, что описанные к настоящему времени мутации обладают различным эффектом. Например, часть мутаций в гене SCN1A, кодирующем α-субъединицу натриевого канала, обусловливают усиление функции канала, что приводит к его длительному открытию, другие ослабляют функцию канала и тем самым затрудняют его открытие [18].
Следует признать, что за редким исключением (например, вигабатрин при синдроме Веста, вызванном туберозным склерозом) специфической высокоэффективной терапии не существует. Тем не менее в публикациях последних лет отмечается определенный опыт по фармокогенетическому подходу к лечению Э.Э. Так, показана наибольшая эффективность лечения при использовании стирипентола при мутациях в гене SCN1A [22], дифенина при мутациях в гене SCN8A [23], леветирацетама при мутациях в гене STXBP1 [24]. В табл. 3 приведены дифференцированные подходы к лечению отдельных синдромов ЭЭ [10].
 Таблица 3. Тактика дифференцированного лечения ЭЭ [10]
Таблица 3. Тактика дифференцированного лечения ЭЭ [10]
Приведенные в данном обзоре данные свидетельствуют о необходимости точной диагностики генетического варианта ранних ЭЭ, что позволит не только осуществить профилактические мероприятия в отягощенной семье, но и повысить эффективность лечения.
Конфликт интересов отсутствует.
1 В данной статье клинические и энцефалографические проявления ЭЭ подробно не рассматриваются, поскольку основное внимание уделяется генетическому аспекту проблемы.